- A+
癌症免疫治疗的发展,特别是免疫检查点阻断疗法,在癌症治疗方面取得了重大突破。然而,仅有不到三分之一的癌症患者能够通过癌症免疫治疗获得显著而持久的治疗效果。
在过去的几十年里,我们了解到,慢性炎症的肿瘤微环境(TME)在肿瘤免疫抑制中起主要作用。而肿瘤相关细胞外基质(ECM)作为TME的核心成员,成为近年来的研究热点。越来越多的研究表明,肿瘤相关ECM是获得更成功的癌症免疫治疗病例的主要障碍之一。
ECM是一种非细胞三维大分子网络,由胶原蛋白、蛋白多糖(PGs)/糖胺聚糖(GAG)、弹性蛋白、纤维连接蛋白(FN)、层粘连蛋白和其他几种糖蛋白组成。无论是在正常组织还是在肿瘤中,基质成分和细胞粘附受体相互结合,形成了一个复杂的网络,其中存在着多种细胞。
多年来,ECM一直被认为是一种惰性的细胞支架,只为细胞提供结构。然而在过去二十年中,人们发现了更多影响细胞生物化学和生物物理过程的功能,ECM被视为生物活性分子的储存库和结合位点。细胞表面受体将信号从ECM传输到细胞,以调节多种细胞功能,如生存、生长、迁移、分化和免疫,这对维持正常内环境平衡至关重要。
大量研究表明,肿瘤相关ECM参与促进肿瘤细胞的生长、侵袭、转移和血管生成,而且抵抗细胞死亡和药物扩散。因此,深入了解ECM与肿瘤免疫反应之间的关系,将有助于发挥靶向肿瘤相关ECM改善癌症免疫治疗的潜力。
细胞外基质是细胞外分泌的大分子(如胶原蛋白、酶和糖蛋白)的复杂网络,其主要功能涉及细胞和组织的结构支架和生化支持。一般而言,ECM可分为基底膜(BM)和间质基质(IM),分别支持上皮/内皮细胞,以及底层基质室和细胞周膜。周围ECM的降解是浸润性癌生长的重要组成部分,更重要的是,ECM的降解伴随着不同肿瘤特异性ECM的沉积,导致密度和硬度的增加。
基底膜由胶原、层粘连蛋白、PGs和FN组成,位于薄壁组织和结缔组织之间的界面,为薄壁细胞提供锚定片状层,以便将其固定在一起,防止其撕裂。在上皮癌中,BM作为癌细胞侵袭、浸润和外渗的结构屏障。在癌症的进展过程中,经常观察到BM的变化,通常癌细胞通过产生ECM重塑酶(如基质金属蛋白酶)、利用BM中的天然孔隙或强行通过这些孔隙侵入BM。
在生理条件下,IM是一种松散的ECM,由胶原蛋白I和III、弹性蛋白纤维和糖蛋白组成,深入BM。成纤维细胞、常驻免疫细胞、血管系统和淋巴管都嵌入其中。然而,在一些肿瘤中,由于胶原纤维沉积增加,IM中的胶原纤维更厚、更有组织、更致密。随着癌症的发展,基质胶原纤维变得越来越整齐,尤其是在肿瘤边缘,从而促进癌细胞的侵袭。
此外,赖氨酰氧化酶(LOX)家族催化胶原交联的形成。在肿瘤中,LOX表达增加导致胶原过度交联,同时,胶原沉积增加,导致硬度增加,从而引起肿瘤中的固体应力。除胶原改变外,ECM中还有各种糖蛋白的病理表达,所有这些都形成一个生态位,促进细胞迁移、粘附和转移。
胶原
胶原蛋白是ECM的主要成分之一,与FN、层粘连蛋白、弹性蛋白和versican等基质糖蛋白一起参与癌症纤维化和实体癌的结构形成。胶原的生物合成始终由成纤维细胞、癌细胞和其他基质细胞(如巨噬细胞)通过多种方式进行调节。ECM中的其他物质,如FN、透明质酸(HA)、层粘连蛋白和基质金属蛋白酶(MMPs),通过整合素、盘状结构域受体(DDR)、酪氨酸激酶受体和一些信号通路与胶原相互作用,从而影响癌细胞的行为和活性。癌症内部基质成分的变化最终形成一个相互反馈回路,影响癌症的预后、复发和治疗抵抗力。
整合素是胶原的主要受体,广泛表达并促进细胞迁移,可能是肿瘤血管生成、化疗抵抗和转移的关键途径。整合素α11β1是一种基质细胞特异性的原纤维胶原受体,在癌相关成纤维细胞(CAF)中过度表达。胶原交联与基质α11表达相关,肿瘤基质α11表达的缺失会引起胶原的重组和硬度降低。β1整合素的强制表达显著刺激Src和细胞外信号调节激酶(ERK)磷酸化,增加细胞硬度并加速细胞运动,导致癌细胞弹性低,转移能力高。整合素还控制ECM和细胞表面TGF-β的局部激活。
此外,非整合素胶原受体DDRs(DDR1和DDR2)在肿瘤细胞表面表达,属于受体酪氨酸激酶(RTK)家族。DDR在与胶原相互作用时表现出延迟和持续的激活,DDR胶原信号在癌症增殖和进展中起着重要作用。
除了上述胶原的作用外,在抗肿瘤免疫中,胶原与肿瘤相关巨噬细胞(TAM)之间的关系也不容低估。TAMs被认为是癌症免疫治疗效果的主要限制因素,而高密度胶原可以引导巨噬细胞获得免疫抑制表型。
蛋白多糖
蛋白聚糖作为ECM的组成部分,在提供协调癌症免疫调节关键事件所需的内在信号方面发挥着关键作用。PGs与癌症相关炎症过程密切相关,并分别调节先天免疫和适应性免疫的关键事件。
Versican是大型硫酸软骨素PGs(CSPG)的hyalectan家族成员,已被证明在许多癌症中过表达。VCAN可通过结合TSG-6和IαI,从循环中释放炎症细胞。此外,VCAN通过HA间接或直接通过CD44、PSGL-1和TLR等受体与炎性细胞相互作用,并激活信号通路以促进炎性细胞因子的合成和分泌,如TNF-α、IL-6和NFκB。
Biglycan(BGN)是小的富含亮氨酸的PGs(SLRPG)的成员,在多种癌症中过表达和分泌,与免疫反应的调节有关。然而,其致癌或抑瘤的潜力尚不清楚。
除VCAN和BGN外,硫酸乙酰肝素蛋白多糖(HSPG)在炎症中也发挥多功能作用,例如调节白细胞募集级联反应的多个步骤,激活淋巴细胞,以及诱导小鼠未成熟树突状细胞(DC)的表型成熟。
糖胺聚糖
透明质酸是一种简单的线性非硫酸化GAG,由N-乙酰氨基葡萄糖(GlcNAc)和葡萄糖醛酸(GlcUA)的重复单元组成,在多种人类实体瘤中集聚。HA的生物活性取决于其分子量和与其相互作用的受体,包括CD44、淋巴内皮受体(LYVE-1)和HA内吞受体(HARE),在正常组织中维持内环境稳定并抑制细胞增殖和迁移。高分子量HA可通过透明质酸酶和自由基裂解成7至200kDa的低分子量(LMW)聚合物,这些LMW-HA可促进炎症、免疫细胞募集和上皮细胞迁移。
上面提到的组分只是数千个ECM组件中的一小部分。它们不仅执行各自的功能,还相互作用,参与ECM和免疫应答的动态变化。
越来越多的研究表明,ECM的重塑在形成肿瘤的炎症和免疫环境中起着重要作用。ECM细胞骨架的重塑、结构可塑性和机械力是免疫突触运输、激活和形成的关键因素。
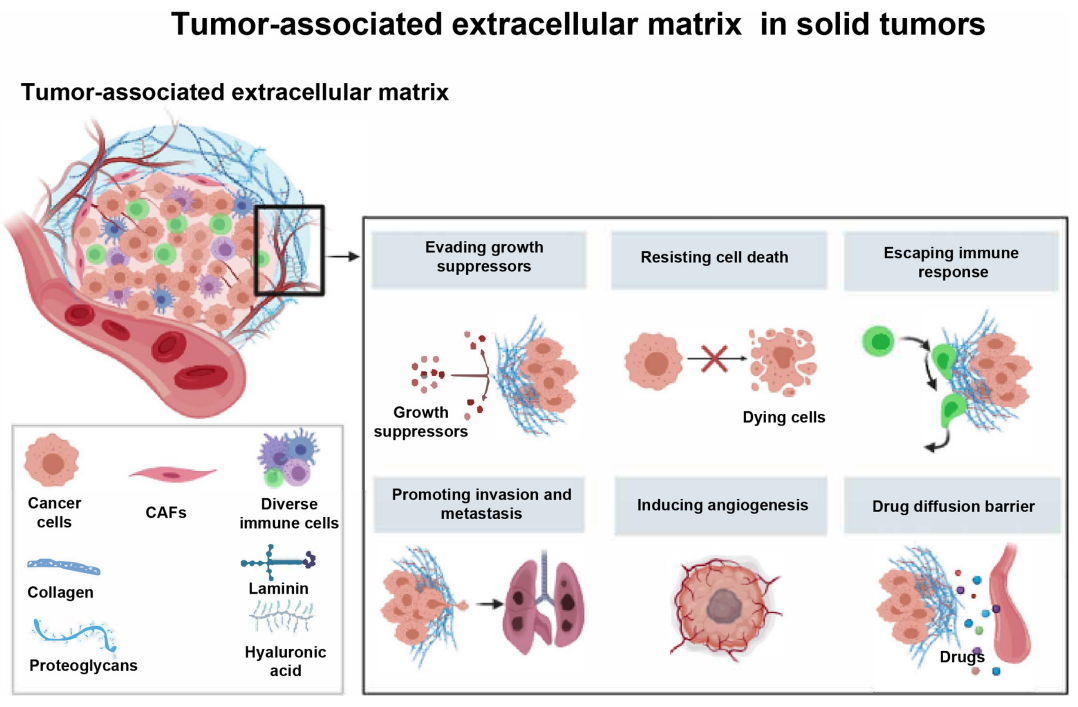
刚性细胞外基质抑制癌细胞死亡并减少抗原的释放
肿瘤细胞外基质的硬度约为周围正常组织的1.5倍,通过在宿主组织上硬化的ECM施加物理力,肿瘤可以增强细胞ECM粘附并打破细胞间接触,从而导致其生长和存活。刚性ECM中可诱导胶原交联,这增强了磷脂酰肌醇3-激酶(PI3K)活性,从而提高癌细胞的生存能力。癌细胞的存活也受到细胞释放MMPs的影响,MMPs可降解ECM的多种成分并与整合素相互作用,从而促进细胞内信号STAT3的活化。此外,ECM还间接参与ERK通路的激活,ERK通路有助于癌细胞的增殖。
ECM除了促进癌细胞的存活外,ECM的硬度也是药物有效摄取或向肿瘤内区域输送的障碍。肿瘤细胞生存潜力的增强这也减少了细胞死亡和癌细胞抗原的释放,作为启动抗癌免疫的第一步和关键步骤,癌细胞抗原释放的减少将削弱癌症免疫。
细胞外基质干扰肿瘤抗原提呈
ICIs反应的功能基础是肿瘤的免疫原性,主要由肿瘤抗原性和抗原呈递效率决定。APC,包括巨噬细胞、DC和B细胞,通过不同机制负责呈递抗原和触发免疫。DC是免疫系统的前哨APC,然而,只有成熟的DC细胞能够诱导抗肿瘤免疫,而未成熟DC细胞呈递的抗原可能导致免疫耐受,并且不能诱导T细胞反应。
值得注意的是,DC功能的最终命运是由微环境的信号决定的,而ECM成分可能诱导具有低免疫原性的DC表型。小鼠髓样DC与层粘连蛋白相互作用的研究表明,小鼠卵巢肿瘤产生多种层粘连蛋白,在这些层粘连蛋白上培养的DC上调AKT和MEK信号通路并降低免疫能力,导致肿瘤生长。
除HS外,HA还可以通过TLR4依赖性方式调节DC的成熟。研究发现,DC暴露于HA片段可增加MHC II、CD80、CD86和CD40等活化标记物的表达,并促进DC活化。此外,HA可以利用VACN形成临时矩阵。HA-VACN相互作用对于炎症细胞的募集非常重要。
细胞外基质影响效应T细胞的启动和激活
一般来说,原始T细胞位于淋巴结(LN)内,与抗原负载的DC相遇并被激活。在LNs中,各种基质细胞亚群形成密集的3D细胞网络,这为幼稚T细胞与DC呈递的抗原相互作用提供了机会,从而引发免疫反应。
在LNs中,免疫或耐受诱导可影响所有基质细胞(SC)中层粘连蛋白α4和α5的表达。在免疫和炎症反应中,层粘连蛋白α5上调;相反,α4在耐受诱导中增加。在功能上,层粘连蛋白411和层粘连蛋白511分别作为CD4+T细胞的共抑制和共刺激配体,层粘连蛋白411抑制CD4+T细胞的激活和Th1、Th2和Th17的极化,但促进Treg极化的诱导。层粘连蛋白511通过α6整合素和α-dystroglycan被CD4+T细胞识别,以抑制T细胞活化、增殖和分化。
细胞外基质调节T细胞的迁移
从LNs到肿瘤部位的效应T细胞对于肿瘤浸润T细胞的密度和多样性至关重要,这与癌症免疫治疗的预后和疗效密切相关。T细胞转运过程是高度动态的,由一系列复杂的机制控制,涉及T细胞和内皮细胞(ECs)之间的复杂相互作用。
T细胞的运输也高度依赖于微环境。T细胞利用多孔三维ECM作为整合素依赖性和受体非依赖性阿米巴运动的支架。层粘连蛋白可以作为配体结合免疫细胞膜受体(主要是整合素)并启动整合素介导的信号传导。
刚性ECM可能作为T细胞浸润肿瘤的物理屏障,并影响T细胞的优先定位。例如,在胰腺导管腺癌(PDAC)和肺癌模型中,基质密度和结构诱导T细胞定位和迁移到肿瘤间质而不是肿瘤细胞巢。
除了刚性外,某些ECM成分也可以在调节T细胞的运动中发挥作用。密集的富含胶原蛋白的ECM对T细胞的浸润和功能有直接和间接的影响。富含胶原纤维的ECM中,CD8+T细胞移动更快、更持久。
细胞外基质干扰T细胞识别和杀死癌细胞
在各种癌症中,胶原纤维比肿瘤间质更厚、更紧密地包裹在癌细胞巢周围。僵硬的ECM可作为肿瘤细胞周围的空间屏障,限制CD8+T细胞的可接近性,从而干扰识别。
癌症的空间分析表明,ECM过度沉积的癌症对免疫检查点抑制具有抵抗力。胶原密度降低肿瘤浸润性T细胞的增殖和杀瘤活性。3D培养T细胞的全转录组分析显示,高密度基质诱导细胞毒性活性标记物(CD101)的下调和Treg标记物(CIP2A)的上调。
此外,肿瘤细胞中PD-L1的表达在逃避“杀伤”步骤中起着重要作用。刚性底物通过肌动蛋白依赖机制增强肺癌细胞中PD-L1的表达,这表明刚性作为肿瘤环境可上调PD-L1的表达,并导致免疫系统逃逸和肿瘤生长。
肿瘤相关ECM可通过多种方式进行治疗靶向,包括靶向ECM分子、ECM重塑酶、改变基质的结构或物理性质,或调节成纤维细胞功能。目前,有多项与ICI的联合研究处于临床阶段。
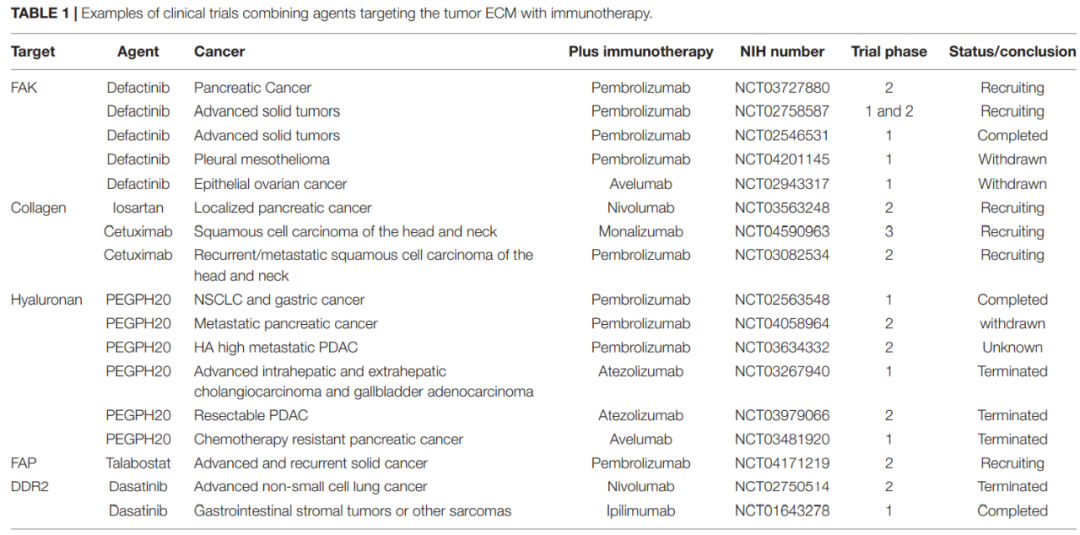
直接靶向
一些研究表明,PG活性调节剂可能是癌症免疫治疗领域的一种新方法。如临床前研究显示,损伤相关分子模式的VCAN片段versikine,有助于骨髓瘤的免疫感应,并增强T细胞激活免疫疗法。非甘氨酸化内聚糖多肽(ESM-1)通过增加体内白细胞浸润和增强先天免疫反应来抑制肿瘤生长。此外,胎盘生长因子-2(PIGF-2)肝素结合域(HBD)偶联免疫检查点抑制剂对多种ECM蛋白表现出极高的亲和力。瘤周注射PIGF-2-抗PD-L1可提高肿瘤组织内的滞留率。除了高效性,它还能降低B16F10黑色素瘤模型的全身毒性。
过量HA的积累可导致间质压力增加,并损害对肿瘤的灌注和化疗。在临床前研究中,聚乙二醇化重组人透明质酸酶α(PEGFH20)已被证明能成功降解肿瘤中的HA并重塑肿瘤基质,从而改善灌注和药物输送。因此,最近的II期HALO-202临床试验(NCT 01839487)表明,在先前未经治疗的转移性PDAC患者中,使用PEGFH20加吉西他滨和Nab紫杉醇治疗可显著提高PFS。然而,III期试验表明,在吉西他滨和Nab紫杉醇中添加PEGFH20可增加ORR,但不能改善OS,这不支持PEGFH20在转移性PDAC中的进一步研究。
透明质酸酶去除HA不仅可以改善化疗的效果,还可以提高免疫治疗的成功率,动物模型已经证明了这一点。此外,透明质酸酶(HAase)通过分解肿瘤ECM中的HA来增加肿瘤组织的通透性,从而增强纳米疫苗诱导的肿瘤特异性T细胞的肿瘤浸润。在存在透明质酸酶的情况下,纳米疫苗和治疗性单克隆抗体的输送都得到了增强。
间接靶向
胶原蛋白是ECM最丰富的成分,主要由成纤维细胞分泌。去除成纤维细胞的研究主要集中在靶向以成纤维细胞激活蛋白(FAP)阳性的成纤维细胞群。在小鼠黑色素瘤模型中,去除表达FAP的CAF可诱导免疫抑制髓系细胞减少。然而,使用小分子抑制剂Talabostat抑制FAP的II期试验未能证明结直肠癌的临床疗效。
除了去除CAF外,其他方法侧重于靶向下游细胞反应以影响ECM。目前,研究的热点主要是基质结合蛋白、整合素及其下游信号机制,如使用小分子激酶抑制剂靶向ECM调节的信号通路,如粘附斑激酶(FAK)和Rho相关蛋白激酶(ROCK)。
越来越多的证据表明,细胞外基质在肿瘤免疫中发挥着极其重要的作用,靶向肿瘤相关ECM具有改善肿瘤免疫治疗的潜力。然而,ECM的成分和结构复杂性以及显著的肿瘤内异质性尚不完全清楚,这可能会限制ECM的靶向治疗的应用。幸运的是,诸如多重免疫组织化学、组织脱细胞技术、单细胞测序和质谱等技术的进步正努力解决上述问题。在未来,调控肿瘤相关ECM的策略有望产生新的方法,进一步优化肿瘤免疫疗法的治疗策略并延长癌症患者的总体生存期。
参考文献:
1.Tumor-Associated Extracellular Matrix: Howto Be a Potential Aide to Anti-tumor Immunotherapy? Front Cell Dev Biol. 2021;9: 739161.
本篇文章来源于微信公众号: 小药说药